Beest Kramer van Santen (BKS)¶
What is it?¶
Began with ab initio calculations of 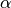 small clusters in a quartz system allows for basis for nearest neighbor interactions also ensures that the force fields is applicable to other polymorphs of the system as they would be made up of the same clusters. At length scales beyond clusters bulk information is taken into account, such as fitting the model to experimental lattice parameters or elastic modulus. Combining both of these approaches has made a model with portability to polymorphs and one that can be extended to other systems.
small clusters in a quartz system allows for basis for nearest neighbor interactions also ensures that the force fields is applicable to other polymorphs of the system as they would be made up of the same clusters. At length scales beyond clusters bulk information is taken into account, such as fitting the model to experimental lattice parameters or elastic modulus. Combining both of these approaches has made a model with portability to polymorphs and one that can be extended to other systems.
It is used for various polymorphs of silica as well as other systems that form tetrahedral network oxides such as AlPo4, Al-O, P-O it can also be used for amorphous silica or carbon
[1]
Equation¶
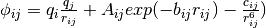
Coulombic term and a covalent (short-range) contribution, into Buckingham form. Coulomb forces are described by the effective silicon charge For charge neutrality
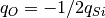
To describe cluster potential energy surface hydrogen atoms can be taken into account, electrostatic interaction 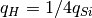
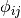 is the interaction energy of atoms i and j
is the interaction energy of atoms i and j
[1]
References¶
[1] Beest, Kramer, S. (1994). Force Fields for Silicas and Aluminophosphates Based on Ab Initio Calculations, 72(20), 3137–3140.