Tersoff¶
What is it?¶
Variation of bond-order potential used to approximate bonding in “different states of silicon” such as liquid, amorphous and diamond silicon. The Brenner potential is an extended version of the Tersoff potential that can be used to model diamond and other phases of carbon. The Tersoff and Brenner models are commonly used to model materials such as Si, Ge, C and alloys of these materials. The total binding energy of the system is the sum of the binding energy for each bond. The eneryg of each bond has a repulsive pairwise contribution and an attractive contribution. The attractive energy is the bond order multiplied by the pairwise energy. The bond order takes into account the local coordination number and the bond angles. The local coordination number being part of the potential allows for different configurations such as linear, trigonal or tertrahedral geometries to be taken into account, which is particularly important for covalent materials. if modelling chemical vapor deposition (CVD) for diamond growth there are additional terms added by Brenner to help describe hydrocarbon bonding. These potentials are called reactive empirical bond order (REBO) potentials. [3]
The Brenner potential allows for accounts for changes in hybridization whends bonds are formed or broken [5].
The Tersoff potential incorporates a dependency on the number of bonds between a pair of atoms. For example, the Tersoff potential would model O-Si-O different from Si-O-Si. The Tersoff potential consists of two-body terms which depend on the local environment. [1]
To fix the issue of overestimating the melting point the Tersoff-ARK potential was developed.
Other varients of the Tersoff potential exist as well such as the ZRL potential which is highly dependent on coordination number in the local envrionment. The effects of a third atom are included which weaken the attractive force in the two-body interaction terms. The Tersoff potential was used in this case due to the flexibility of its short range interactions and success in modelling condensed phases. For ZRL the Tersoff potential was modified to include the electrostatic potential and a self energy term. [2]
For additional information view [6]
Well-Described¶
The Tersoff potential can well describe properties of liquid and amorphous phases of silicon.
ZRL better describes mixed systems and accounts for doordination defects. It is good for systems involving Si, N, O, H in noncrystalline phases. Can be used to model large-scale complex heterogeneous systems in combination with DFT based MD [2]
Issues¶
The Tersoff potential predicts the melting temperature of silicon to be higher than the actual value.
Tersoff and Brenner potentials may not correctly model the angular dependency of 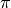 bonds. However, there has been work to improve this by adding terms with angular depenencies [4]
bonds. However, there has been work to improve this by adding terms with angular depenencies [4]
Equation¶
The general form of the Tersoff equation is given by
![\phi_{ij}(r_{ij})= [\phi_{R}(r_{ij})-B_{ij}\phi_{A}(r_{ij})]](../_images/math/4d9f4bb6d7ffc08d2cacbcedf94c16e129166130.png)
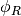 and
and 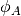 are repulsive and attractive potentials
are repulsive and attractive potentials
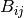 is the bond order between
is the bond order between 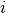 and
and 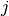 , as the bond coordination increases
, as the bond coordination increases 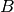 will decrease
will decrease
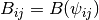
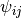 is a function describing the number of neighbor atoms within a certain distance and the angles between the atoms, which describes how the atoms are bonded together.
[3]
is a function describing the number of neighbor atoms within a certain distance and the angles between the atoms, which describes how the atoms are bonded together.
[3]
Silicon potential function from Tersoff
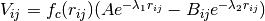
Repulsive term - attractive term
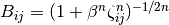
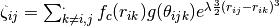
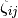 describes angular effects, it depends on the number of pair potential bonds in the summation. effective coordination number of atom [6]
takes into account the relative distance between two neighboring atoms
describes angular effects, it depends on the number of pair potential bonds in the summation. effective coordination number of atom [6]
takes into account the relative distance between two neighboring atoms 
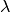 is the bond strength
is the bond strength
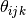 is the angle between bonds
is the angle between bonds 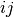 and
and 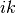
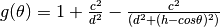
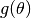 will be minimized when
will be minimized when 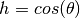
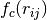 is a cut-off radius function
is a cut-off radius function
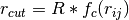
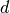 determines how “sharp” the angular dependency is
determines how “sharp” the angular dependency is
 is the strength of the angular effect
is the strength of the angular effect
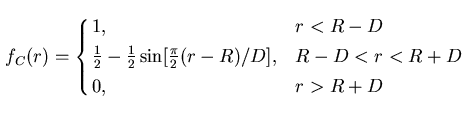
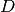 is the layer thickness
is the layer thickness
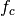 decreases from 1 to 0 in the range
decreases from 1 to 0 in the range 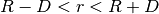
[5],[6]
Plot¶
Examples of Use¶
In [1] parameters set developed for Tersoff which predicts well propeerties of SiO2 such as phase transition pressures, structural parameters, density and cohesive energy, this paper neglects Coulombic interactions
References¶
[1] Munetoh, S., Motooka, T., Moriguchi, K., & Shintani, A. (2007). Interatomic potential for Si-O systems using Tersoff parameterization. Computational Materials Science, 39(2), 334–339. http://doi.org/10.1016/j.commatsci.2006.06.010
[2] Billeter, S. R., Curioni, A., Fischer, D., & Andreoni, W. (2006). Ab initio derived augmented Tersoff potential for silicon oxynitride compounds and their interfaces with silicon. Physical Review B - Condensed Matter and Materials Physics, 73(15), 1–15. http://doi.org/10.1103/PhysRevB.73.155329
[3] LeSar, Richard. Introduction to computational materials science: fundamentals to application s. Cambridge University Press, 2013.
[4] Pettifor, D. G., & Oleynik, I. I. (2004). Interatomic bond-order potentials and structural prediction. Progress in Materials Science, 49(3–4), 285–312. http://doi.org/10.1016/S0079-6425(03)00024-0
[5] Pettifor, D. G., & Oleynik, I. I. (2004). Interatomic bond-order potentials and structural prediction. Progress in Materials Science, 49(3–4), 285–312. http://doi.org/10.1016/S0079-6425(03)00024-0